Introduction
Overview
Teaching: 0 min
Exercises: 0 minQuestions
What is Common Workflow Language?
How are CWL workflows written?
How do CWL workflows compare to shell workflows?
What are the advantages of using CWL workflows?
Objectives
First learning objective. (FIXME)
Computational workflows are widely used for data analysis, enabling rapid innovation and decision making. Workflow thinking is a form of “conceptualizing processes as recipes and protocols, structured as [work- or] dataflow graphs with computational steps, and subsequently developing tools and approaches for formalizing, analyzing and communicating these process descriptions” (Gryk & Ludascher, 2017).
However as the rise in popularity of workflows has been matched by a rise in the number of dispirit workflow managers that are available, each with their own standards for describing the tools and workflows, reducing portability and interoperability of these workflows.
CWL is a free and open standard for describing command-line tool based workflows. These standards provide a common, but reduced, set of abstractions that are both used in practice and implemented in many popular workflow systems. The CWL language is declarative, enabling computational workflows to be constructed from diverse software tools, executing each through their command-line interface.
Previously researchers might write shell scripts to link together these command-line tools. Although these scripts might provide a direct means of accessing the tools, writing and maintaining them requires specific knowledge of the system that they will be used on. Shell scripts are not easily portable, and so researchers can easily end up spending more time maintaining the scripts than carrying out their research. The aim of CWL is to reduce that barrier of usage of these tools to researchers.
CWL workflows are written in a subset of YAML, with a syntax that does not restrict the amount of detail provided for a tool or workflow. The execution model is explicit, all required elements of a tool’s runtime environment must be specified by the CWL tool-description author. On top of these basic requirements they can also add hints or requirements to the tool-description, helping to guide users (and workflow engines) on what resources are needed for a tool.
The CWL standards explicitly support the use of software container technologies, helping ensure that the execution of tools is reproducible. Data locations are explicitly defined, and working directories kept separate for each tool invocation. These standards ensure the portability of tools and workflows, allowing the same workflows to be run on your local machine, or in a HPC or cloud environment, with minimal changes required.
Key Points
First key point. Brief Answer to questions. (FIXME)
CWL and Shell Tools
Overview
Teaching: 0 min
Exercises: 0 minQuestions
What is the difference between a CWL tool description and a CWL workflow?
How can we create a tool descriptor?
How can we use this in a single step workflow?
Objectives
describe the relationship between a tool and its corresponding CWL document
exercise good practices when naming inputs and outputs
understand how to reference files for input and output
explain that only files explicitly mentioned in a description will be included in the output of a step/workflow
implement bulk capturing of all files produced by a step/workflow for debugging purposes
use STDIN and STDOUT as input and output
capture output written to a specific directory, the working directory, or the same directory where input is located
learning objectives
By the end of this episode, learners should be able to explain how a workflow document describes the input and output of a workflow and the flow of data between tools and describe all the requirements for running a tool and define the files that will be included as output of a workflow.
CWL workflows are defined in a YAML script, containing the workflow and the requirements for running that workflow. All CWL scripts should start with two lines of code:
cwlVersion: v1.2
class:
The cwlVersion string defines which standard of the language is required for the tool or workflow. The most recent version is v1.2, and defaulting to this will enable your scripts to use all versions of the language, though some workflow engines which are not up-to-date may not run the script. This is, however, a hurdle to be tackled when you reach it.
The class field defines what this particular script is. The majority of CWL scripts will fall into one of two classes: CommandLineTool, or Workflow. The former class is used for describing the interface for a command-line tool, while the latter class is used for collecting those tool descriptions into a workflow. In this lesson we will learn the differences between these two classes, how to pass data to and from command-line tools and specify working environments for these, and finally how to use a tool descriptor within a workflow.
Tool Descriptors
To demonstrate the basic requirements for a tool descriptor we will recreate the standard hello world example. Note: replace this with a domain specific (but similar complexity) example! This is the shell echo command that we will use:
$ echo 'hello World!'
hello World!
Create a file, echo.cwl, to contain your CWL example for this:
cwlVersion: v1.2
class: CommandLineTool
baseCommand: [echo, 'hello World!']
We present baseCommand as a two item list containing the command and the input string. CWL will combine these two items (in the order given) to make the full command when the script is run. This is not a complete tool descriptor yet, however - to find out what is missing we can use cwl-runner to validate the script:
$ cwl-runner --validate echo.cwl
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
ERROR Tool definition failed validation:
echo.cwl:1:1: "outputs" section is not valid.
cwl-runner has noted that we are missing an outputs section, so we will add this to our script:
cwlVersion: v1.2
class: CommandLineTool
baseCommand: [echo, 'hello World!']
outputs: []
note that we are using an empty list [] here, as we do not want to capture any output for the moment. We will now validate the script again:
$ cwl-runner --validate echo.cwl
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
ERROR Tool definition failed validation:
echo.cwl:1:1: Object `echo.cwl` is not valid because
tried `CommandLineTool` but
missing required field `inputs`
cwl-runner has noted that we are missing an inputs field, so we will add this also to our script (again as an empty list):
cwlVersion: v1.2
class: CommandLineTool
baseCommand: [echo, 'hello World!']
inputs: []
outputs: []
We will now validate the script again:
$ cwl-runner --validate echo.cwl
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
echo.cwl is valid CWL.
Finally we have a valid CWL tool descriptor, so we will run this using cwl-runner:
cwl-runner echo.cwl
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
INFO [job echo.cwl] /private/tmp/docker_tmpwvj2kdvw$ echo \
'hello World!'
hello World!
INFO [job echo.cwl] completed success
{}
INFO Final process status is success
Our script has been run successfully! Note: add graphic here that identifies what each part of the returned information is (for example, what is CWL information, what is the command run, what is STDOUT, and what is the (currently empty) returned information
Location, Location, Location
Note the string after
INFO [job echo.cwl]shows the location/private/tmp/docker_tmpwvj2kdvwin our example, but will show a different randomly named, and temporary, directory when you run this example. CWL will always create a separate, temporary, working directory for running each tool instance. This ensures that the run-time environment for each tool instance is well controlled, and does not contain anything left behind by another tool.
Script order
To make our script more readable we have put the
inputfield in front of theoutputfield. However CWL syntax requires only that each field is properly defined, it does not require them to be in a particular order. Try changing around the order of fields in our example script, and run the validation step on these to make sure this is true.
So far our script is rather limited, with no inputs specified, and the string that we are printing out has been merged into the baseCommand. We will now split out the input string, so that we can make this tool more flexible.
We remove the hello World! string from the baseCommand (where it should not have been in the first place…), and create an inputs item which we will call message_text:
cwlVersion: v1.2
class: CommandLineTool
baseCommand: echo
inputs:
message_text:
type: string
default: 'hello World!'
inputBinding:
position: 1
outputs: []
We set the type of message_text to string, and set the inputBinding position (defining where the input item appears after the baseCommand) as 1. We also give a default value, hello World!, for this item, which will be used if the item is not defined within an input file.
We can now validate, and then run, this tool again:
$ cwl-runner --validate echo.cwl
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
echo.cwl is valid CWL.
$ cwl-runner echo.cwl
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
INFO [job echo.cwl] /private/tmp/docker_tmprm65mucw$ echo \
'hello World!'
hello World!
INFO [job echo.cwl] completed success
{}
INFO Final process status is success
The script is now ready to accept an input from us. This we will put in another YAML file (moon.yml):
message_text: 'hello Moon!'
And then run:
$ cwl-runner echo.cwl moon.yml
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
INFO [job echo.cwl] /private/tmp/docker_tmpu7z20wc7$ echo \
'hello Moon!'
hello Moon!
INFO [job echo.cwl] completed success
{}
INFO Final process status is success
Note that we have not yet captured the output from the command. We can do this by adding an outputs item to the script:
cwlVersion: v1.2
class: CommandLineTool
baseCommand: echo
inputs:
message_text:
type: string
default: 'hello World!'
inputBinding:
position: 1
outputs:
message_out:
type: stdout
Here we have added the message_out item, which has been given type stdout (as we want to capture the output at the command line, rather than any particular files).
Now we run the script:
$ cwl-runner echo.cwl moon.yml
INFO .../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
INFO [job echo.cwl] /private/tmp/docker_tmp7k0bqeg5$ echo \
'hello Moon!' > /private/tmp/docker_tmp7k0bqeg5/9611f21693018fe4ce4bf1f3884e47dae2ce3aab
INFO [job echo.cwl] completed success
{
"message_out": {
"location": "file:///.../9611f21693018fe4ce4bf1f3884e47dae2ce3aab",
"basename": "9611f21693018fe4ce4bf1f3884e47dae2ce3aab",
"class": "File",
"checksum": "sha1$d4413a97a36059e8855168ac7939a4cb5d4da9c9",
"size": 12,
"path": "/.../9611f21693018fe4ce4bf1f3884e47dae2ce3aab"
}
}
INFO Final process status is success
Note that the output has been saved as a file called 9611f21693018fe4ce4bf1f3884e47dae2ce3aab (not message_out, which is only the output variable name) in this instance. When you run this script the file name will be different. You can open this text file with a text editor to confirm that it contains the expected message.
It is not very user-friendly to have our script return a randomly named file each time, so we will make use of the stdout field to specify the name of the text file that we want standard output to be captured to:
cwlVersion: v1.2
class: CommandLineTool
baseCommand: echo
inputs:
message_text:
type: string
default: 'hello World!'
inputBinding:
position: 1
stdout: output.txt
outputs:
message_out:
type: stdout
Running the script will produce this output:
$ cwl-runner echo.cwl moon.yml
INFO /.../cwl-runner 3.0.20200807132242
INFO Resolved 'echo.cwl' to 'file:///.../echo.cwl'
INFO [job echo.cwl] /private/tmp/docker_tmp5iazb06g$ echo \
'hello Moon!' > /private/tmp/docker_tmp5iazb06g/output.txt
INFO [job echo.cwl] completed success
{
"message_out": {
"location": "file:///.../output.txt",
"basename": "output.txt",
"class": "File",
"checksum": "sha1$d4413a97a36059e8855168ac7939a4cb5d4da9c9",
"size": 12,
"path": "/.../output.txt"
}
}
INFO Final process status is success
Note now that the file returned is called output.txt, but it has the same contents as the previous, randomly named, file.
We will now make use of the VSCode/Benten tool to illustrate the tool descriptor. Please press the CWL Viewer button at the top-left of the VSCode window:
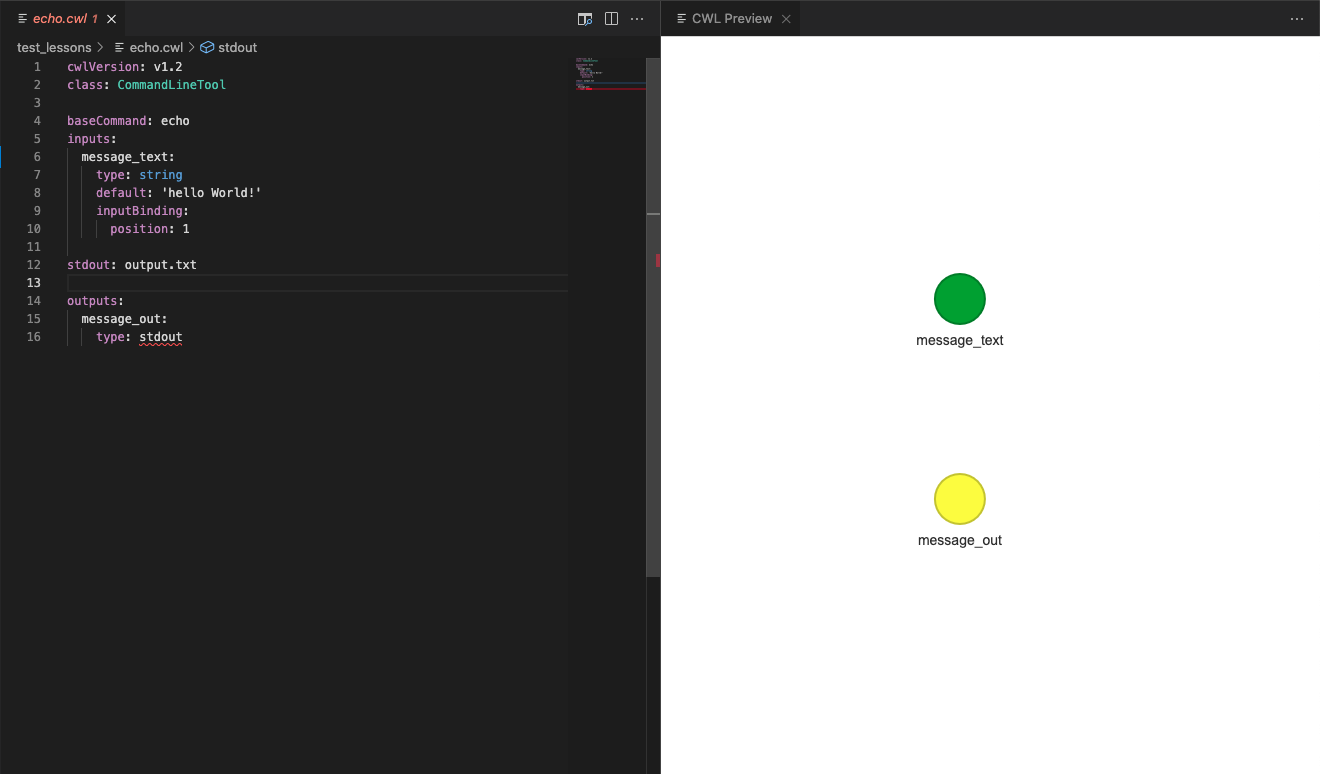
You will now be able to see a sketch of the tool descriptor. This will show the input (in green) and output (in yellow) items for this tool descriptor. This is not very helpful information at the moment, but leave this window open as we move onto writing our first workflow.
Creating tool descriptor for your tool
Add here a challenge for creating a tool descriptor for another tool? Perhaps make the input a file and a text string, and introduce
prefix: ...to theinputBindinginterface (as well as giving more positions than just 1)?
CWL single step workflow
Now that we have created a tool descriptor we can use it in our workflow. We will start with a single step workflow, to illustrate how workflows and tool descriptors interact.
Create a file workflow_example.cwl, containing these lines:
cwlVersion: v1.2
class: Workflow
inputs: []
outputs: []
Workflows use inputs and outputs fields, just as the tool descriptors do, but they don’t use baseCommand. Run the validation tool to find out what is missing:
$ cwl-runner --validate workflow_example.cwl
INFO /.../cwl-runner 3.0.20200807132242
INFO Resolved 'workflow_example.cwl' to 'file:///.../workflow_example.cwl'
ERROR Tool definition failed validation:
workflow_example.cwl:1:1: Object `workflow_example.cwl` is not valid because
tried `Workflow` but
missing required field `steps`
Workflows need a steps field, in which are listed the workflow tasks or steps that are to be run. In this instance we wish only to run the echo.cwl tool that we wrote above, so we will add one step to this workflow. This step will require us to specify what tool we will run, as well as providing lists of in and out items for the tool. To begin with we will provide the bare minimum to make this workflow run:
cwlVersion: v1.2
class: Workflow
inputs: []
outputs: []
steps:
01_echo:
run: echo.cwl
in: []
out: []
And then we run the script:
$ cwl-runner workflow_example.cwl
INFO /.../cwl-runner 3.0.20200807132242
INFO Resolved 'workflow_example.cwl' to 'file:///.../workflow_example.cwl'
INFO [workflow ] start
INFO [workflow ] starting step 01_echo
INFO [step 01_echo] start
INFO [job 01_echo] /private/tmp/docker_tmpx4889wo6$ echo \
'hello World!' > /private/tmp/docker_tmpx4889wo6/output.txt
INFO [job 01_echo] completed success
INFO [step 01_echo] completed success
INFO [workflow ] completed success
{}
INFO Final process status is success
This was a success, but the workflow has not returned any files this time, and the echo’d message is the default ‘hello World!’ message. Now we must connect our tool inputs and outputs up in the workflow.
First we will specify the flow of inputs for our workflow, taking them from the YAML configuration file, and passing them through to the echo tool:
cwlVersion: v1.2
class: Workflow
inputs:
message_text: string
outputs: []
steps:
01_echo:
run: echo.cwl
in:
message_text: message_text
out: []
The inputs entry is similar to that for the echo.cwl tool (as we are going to read the same input file), but we have not given a default value or input binding. Within the in list we explicitly link the tool’s message_text field with our workflow’s message_text field. These do not need to have matching names, in the next episode we will show how these can change as you start linking steps together.
Now run this workflow:
$ cwl-runner workflow_example.cwl moon.yml
INFO /.../cwl-runner 3.0.20200807132242
INFO Resolved 'workflow_example.cwl' to 'file:///.../workflow_example.cwl'
INFO [workflow ] start
INFO [workflow ] starting step 01_echo
INFO [step 01_echo] start
INFO [job 01_echo] /private/tmp/docker_tmpd9ghguo8$ echo \
'hello Moon!' > /private/tmp/docker_tmpd9ghguo8/output.txt
INFO [job 01_echo] completed success
INFO [step 01_echo] completed success
INFO [workflow ] completed success
{}
INFO Final process status is success
We can see that the echo’d text has changed. But still no files are being returned from our tool, so we need to explicitly list the files that we want the workflow to return.
cwlVersion: v1.2
class: Workflow
inputs:
message_text: string
outputs:
message_file:
type: File
outputSource: 01_echo/message_out
steps:
01_echo:
run: echo.cwl
in:
message_text: message_text
out: [message_out]
The outputs entry is again similar to that for the echo.cwl tool. However we are using the File type, as that is what the tool descriptor returns, and specifying an outputSource, which makes clear the link to step 01_echo and the message_out object. For the out field of our workflow step we simply provide a list of the objects we require outputting.
Now we can run this workflow, to provide the same output as running the tool descriptor did:
$ cwl-runner workflow_example.cwl moon.yml
INFO /.../cwl-runner 3.0.20200807132242
INFO Resolved 'workflow_example.cwl' to 'file:///.../workflow_example.cwl'
INFO [workflow ] start
INFO [workflow ] starting step 01_echo
INFO [step 01_echo] start
INFO [job 01_echo] /private/tmp/docker_tmpvkqoq0n3$ echo \
'hello Moon!' > /private/tmp/docker_tmpvkqoq0n3/output.txt
INFO [job 01_echo] completed success
INFO [step 01_echo] completed success
INFO [workflow ] completed success
{
"message_file": {
"location": "file:///.../output.txt",
"basename": "output.txt",
"class": "File",
"checksum": "sha1$d4413a97a36059e8855168ac7939a4cb5d4da9c9",
"size": 12,
"path": "/.../output.txt"
}
}
INFO Final process status is success
In the CWL Preview window of the VSCode editor we will now be able to see the input and outputs that were there for the tool descriptor, but these will now be connected by the step of our workflow, illustrating their connection.
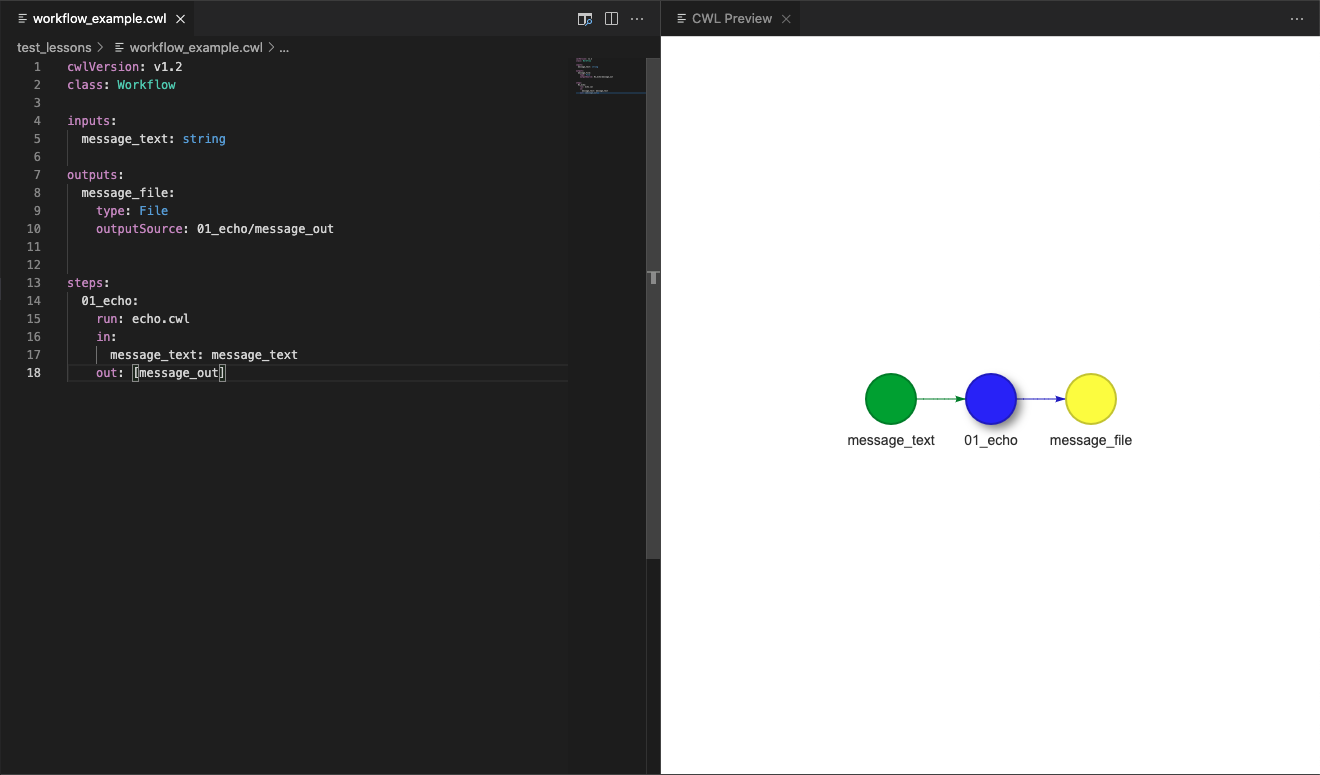
This connection is quite basic here, but in the next episode we will make use of this feature of VSCode/Benten to plan more complicated workflows.
Example Exercises
Use https://github.com/bcosc/fast_genome_variants/blob/main/README.md to create a CommandLineTool
Exercise 1
Create the baseCommand for running the joint_haplotype caller using the fast_genome_variants README.
Solution
The base command should use the path to the binary and the type of variants you’re calling.
baseCommand: [fgv, joint_haplotype]
Exercise 2:
When working in a cloud environment, you need to specify what machine type you would like to run on. Which means the job has to have specific parameters describing the RAM, Cores and Disk space (for both temporary and output files) it requires.
Create the
ResourceRequirementsfield for running 2 BAMs for thefgv joint_haplotypecommand.Solution:
requirements: ResourceRequirement: ramMin: 4000 coresMin: 2FGV requires 2 GiB of memory for each bam input, and the unit for
ramMinis in MiB, so we need approximately 4000 MiB to meet the requirement. FGV also requires 1 core for each BAM, so here we ask for at least 2 cores.
Exercise 3:
- Create the
inputfield for runningfgv_joint_haplotype- Add an optional flag for calling a GVCF output
- Add a string input for intervals
chr2:1-10000- Add an output name.
Solution:
inputs: bam: type: File[] inputBinding: position: 1 prefix: -bam secondaryFiles: - .bai gvcf: type: boolean inputBinding: position: 2 prefix: -gvcf interval: type: string inputBinding: position: 3 output_name: type: string inputBinding: position: 4 prefix: -o
Exercise 4:
Create the output variable for the CommandLineTool and name it output_vcf.
Solution:
outputs: output_vcf: type: File outputBinding: glob: $(inputs.output_name)
Exercise 5:
TODO
Solution:
Capturing Output
Exercise 1
Using this
CommandLineTooldescription, what files would be the output of the tool?cwlVersion: v1.0 class: CommandLineTool baseCommand: [bwa, mem] inputs: reference: type: File inputBinding: position: 1 fastq_reads: type: File[] inputBinding: position: 2 stdout: output.sam outputs: output: type: File outputBinding: glob: output.samSolution
output.samwill be the only file outputted. Only files explicitly stated in the outputs field will be included in the output of the step.
Exercise 2
Your colleague tells you to run
fastqc, which creates several files describing the quality of the data. For now, let’s assume the tool creates three files:
final_report_fastqc.htmlfinal_figures_fastqc.zipsupplemental_figures_fastqc.htmlCreate a CWL
outputsfield using aFilearray that captures all the fastqc files in a single output variable.Solution
outputs: output: type: File[] outputBinding: glob: "*fastqc*"Actually,
fastqcmay create more than 3 of these files, depending on the input parameters you give it, it may create aresultsdirectory that contains additional files such asresults/fastqc_per_base_content.htmlandresults/fastqc_per_base_gc_content.html.Create a CWL
outputsfield that captures theresults/fastqc_per_base_content.htmlandresults/fastqc_per_base_gc_content.htmlin separate output variables.Solution
outputs: per_base_content: type: File outputBinding: glob: "results/fastqc_per_base_content.html" per_base_gc_content: type: File outputBinding: glob: "results/fastqc_per_base_gc_content.html"Finally, instead of explicitly defining each file to be captured, create a CWL
outputsfield that captures the entireresultsdirectory.Solution
outputs: results: type: Directory outputBinding: glob: "results"
Exercise 3
Since
fastqccan be unpredictable in its outputs and file naming, create a CWL outputs field using aDirectorythat captures all the files in a single output variable.Solution
outputs: output: type: Directory outputBinding: glob: .
Exercise 4
Your colleague says that he is running
samtools indexin CWL, but the index is not being outputted. Fix the following CWL to have output the index along with thebamas asecondaryFile.cwlVersion: v1.0 class: CommandLineTool requirements: InitialWorkDirRequirement: listing: - $(inputs.bam) baseCommand: [samtools, index] inputs: bam: type: File inputBinding: position: 1 valueFrom: $(self.basename) outputs: output_bam_and_index: type: File outputBinding: glob: $(inputs.bam.basename)Solution
cwlVersion: v1.0 class: CommandLineTool requirements: InitialWorkDirRequirement: listing: - $(inputs.bam) baseCommand: [samtools, index] inputs: bam: type: File inputBinding: position: 1 valueFrom: $(self.basename) outputs: output_bam_and_index: type: File secondaryFiles: - .bai outputBinding: glob: $(inputs.bam.basename)
Exercise 5
What if
InitialWorkDirRequirementwas not used, and the index file was created where the input bam was located? How would you capture the output? Create theoutputsfield using the same CWL in exercise 4.Solution
outputs: output_bam_and_index: type: File secondaryFile: - .bai outputBinding: glob: $(inputs.bam)
Key Points
First key point. Brief Answer to questions. (FIXME)
Developing Multi-Step Workflows
Overview
Teaching: 0 min
Exercises: 0 minQuestions
How can we expand to a multi-step workflow?
Iterative workflow development
Workflows as dependency graphs
How to use sketches for workflow design?
Objectives
graph objectives:
explain that a workflow is a dependency graph
sketch objectives:
use cwlviewer online
generate Graphviz diagram using cwltool
exercise with the printout of a simple workflow; draw arrows on code; hand draw a graph on another sheet of paper
iterate objectives:
recognise that workflow development can be iterative i.e. that it doesn’t have to happen all at once
By the end of this episode, learners should be able to explain that a workflow is a dependency graph and sketch their workflow, both by hand, and with an automated visualizer and recognise that workflow development can be iterative i.e. that it doesn’t have to happen all at once.
Key Points
First key point. Brief Answer to questions. (FIXME)
Resources for Reusing Tools and Scripts
Overview
Teaching: 0 min
Exercises: 0 minQuestions
How to find other tools/solutions for awkward problems?
Objectives
tools objectives:
know good resources for finding solutions to common problems
By the end of this episode, learners should be aware of where they can look for CWL recipes and more help for common, but awkward, tasks.
Key Points
First key point. Brief Answer to questions. (FIXME)
Documentation and Citation in Workflows
Overview
Teaching: 0 min
Exercises: 0 minQuestions
How to document your workflow?
How to cite research software in your workflow?
Objectives
Documentation Objectives:
explain the importance of documenting a workflow
use description fields to document purpose, intent, and other factors at multiple levels within their workflow
recognise when it is appropriate to include this documentation
Citation Objectives:
explain the importance of correctly citing research software
give credit for all the tools used in their workflow(s)
By the end of this episode, learners should be able to document their workflows to increase reusability and explain the importance of correctly citing research software.
TODO (CITE): define some specific objectives to capture the skills being taught in this section.
See this page.
Finding an identifier for the tool
(Something about permanent identifiers insert here)
When your workflow is using a pre-existing command line tool, it is good practice to provide citation for the tool, beyond which command line it is executed with.
The SoftwareRequirement hint can list named packages that should be installed in order to run the tool.
So for instance if you installed using the package management system with apt install bamtools the package bamtools can be
cited in CWL as:
hints:
SoftwareRequirement:
packages:
bamtools: {}
Adding version
Q: bamtools --version prints out blablabla 2.3.1 - how would you indicate in CWL that this is the version of BAMTools the workflow was tested against?
A:
hints:
SoftwareRequirement:
packages:
bamtools:
version: ["2.3.1"]
Adding Permanent identifiers
To help identify the tool across package management systems we can also add permanent identifiers and URLs, for instance to:
- RRID to SciCrunch
- bio.tools registration
- DOI to a publication
- Homepage
- source repository (e.g. GitHub)
These can be added to the specs list:
hints:
SoftwareRequirement:
packages:
interproscan:
specs: [ "https://identifiers.org/rrid/RRID:SCR_005829" ]
version: [ "5.21-60" ]
How to find a RRID permanent identifier
RRID provides identifiers for many commonly used resources tools in bioinformatics. For instance, a search for BAMtools finds an entry for BAMtools with identifier RRID:SCR_015987 and additional information.
We can transform the RRID into a Permanent Identifier (PID) for use in CWL using http://identifiers.org/ by appending the RRID to https://identifiers.org/rrid/ - making the PID https://identifiers.org/rrid/RRID:SCR_015987 which we see resolve to the same SciCrunch entry, and add to our specs list:
hints:
SoftwareRequirement:
packages:
interproscan:
specs: [ "https://identifiers.org/rrid/RRID:SCR_015987" ]
Note that as CWL is based on YAML we use "quotes" to escape these identifiers include the : character.
Finding bio.tools identifiers
As an alternative to RRID we can add identifiers from the ELIXIR Tools Registry https://bio.tools/ - for instance https://bio.tools/bamtools
hints:
SoftwareRequirement:
packages:
bamtools:
specs:
- "https://identifiers.org/rrid/RRID:SCR_015987"
- "https://bio.tools/bamtools"
- How to write a DOI as a PID URI https://www.nature.com/articles/nmeth.1923 -> https://doi.org/ + 10.1038/nmeth.1923 -> https://doi.org/10.1038/nmeth.1923
Package manager identifiers
Q: You have used apt install bamtools in the Linux distribution Debian 10.8 “Buster”. How would you in CWL SoftwareRequirement identify the Debian package recipe, and with which version?
A:
hints:
SoftwareRequirement:
packages:
bamtools:
specs:
- "https://identifiers.org/rrid/RRID:SCR_015987"
- "https://bio.tools/bamtools"
- "https://packages.debian.org/buster/bamtools"
version: ["2.5.1", "2.5.1+dfsg-3"]
This package repository has a URI for each installable package, depending on the distribution, we here pick "buster". While the upstream GitHub repository of bamtools has release version v2.5.1, the Debian packaging adds +dfsg-3 to indicate the 3rd repackaging with additional patches, in this case to make the software comply with Debian Free Software Guidelines (dfsg).
Under version list in CWL we’ll include 2.5.1 which is the upstream version, ignoring everything after + or - according to semantic versioning rules. As an optional extra you can also include the Debian-specific version "2.5.1+dfsg-3" to indicate which particular packaging we tested the workflow with at the time.
Exercise: There is a “obvious” DOI
Q: You have a workflow using bowtie2, how would you add a citation?
A:
hints:
SoftwareRequirement:
packages:
bowtie2:
specs: [ "https://doi.org/10.1038/nmeth.1923" ]
version: [ "1.x.x" ]
RRID for bowtie2
RRID:SCR_005476 -> https://scicrunch.org/resolver/RRID:SCR_005476 #bowtie not bowtie2 https://identifiers.org/rrid/ + RRID -> https://identifiers.org/rrid/RRID:SCR_005476 PID
https://bio.tools/bowtie2
http://bioconda.github.io/recipes/bowtie2/README.html vs. https://anaconda.org/bioconda/bowtie2
Giving clues to reader
Authorship/citation of a tool vs the CWL file itself (particularly of a workflow)
Add identifiers under requirements? https://www.commonwl.org/user_guide/20-software-requirements/index.html
SciCrunch - looking up RRID for Bowtie2 Then bio.tools
hints:
SoftwareRequirement:
packages:
interproscan:
specs: [ "https://identifiers.org/rrid/RRID:SCR_005829",
"http://somethingelse"]
version: [ "5.21-60" ]
Trickier: Only Github and homepage
s:codeRepository:
hints:
SoftwareRequirement:
packages:
interproscan:
specs: [ "https://github.com/BenLangmead/bowtie2"]
version: [ "fb688f7264daa09dd65fdfcb9d0f008a7817350f" ]
No version, add commit ID or date instead as version
–> (How to make Your own tool citable?)
Getting credit for your CWL files
NOTE: Difference between credit for this CWL file vs credit for the tool it calls.
s:author "Me"
s:dateModified: "2020-10-6"
s:version: "2.4.2"
s:license: https://spdx.org/licenses/GPL-3.0
https://www.commonwl.org/user_guide/17-metadata/index.html
Using s:citation?
something like..
s:citation: https://dx.doi.org/10.1038/nmeth.1923
s:url: http://example.com/tools/
s:codeRepository: https://github.com/BenLangmead/bowtie2
$namespaces:
s: https://schema.org/
$schemas:
- http://schema.org/version/9.0/schemaorg-current-http.rdf
—> Need new guidance on how to publish workflows, making DOIs in Zenodo, Dockstore etc. https://docs.bioexcel.eu/cwl-best-practice-guide/devpractice/publishing.html https://guides.github.com/activities/citable-code/
How to do it properly to improve findability.
How to publisize CWL tools
CWL workflow descriptions
About how to wire together CommandLineTool steps in a cwl Workflow file.
Key Points
First key point. Brief Answer to questions. (FIXME)
Debugging Workflows
Overview
Teaching: 0 min
Exercises: 0 minQuestions
introduce within above lessons?
Objectives
interpret commonly encountered error messages
solve these common issues
By the end of this episode, learners should be able to recognize and fix simple bugs in their workflow code.
(non-exhaustive) list of possible examples:
- YAML errors
- “wiring errors” e.g. where is the output from my step?
- type mismatch
- array vs single-item mismatch
- no formats on input but format is required by workflow
Key Points
First key point. Brief Answer to questions. (FIXME)